021-67658806
在线订单
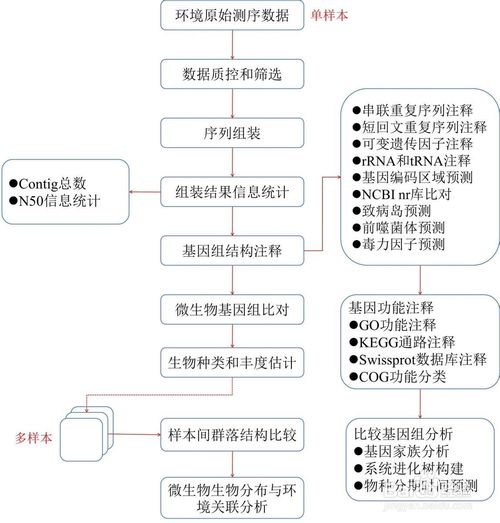
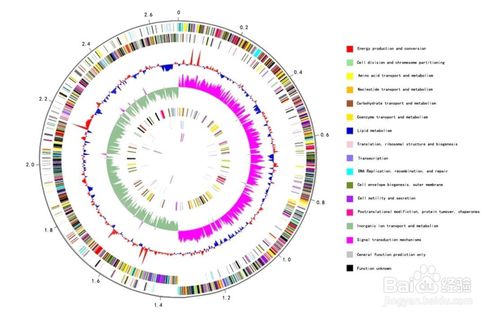
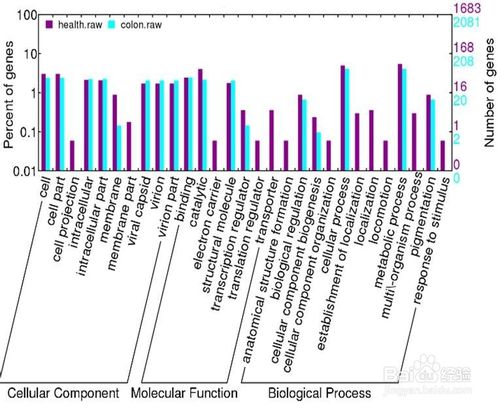
宏基因组 ( Metagenome)(也称微生物环境基因组Microbial Environmental Genome, 或元基因组)是由 Handelsman 等 1998 年提出的新名词, 其定义为“the genomes of the total microbiota found in nature” , 即环境中全部微小生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因,目前主要指环境样品中的细菌和真菌的基因组总和。宏基因组学(或元基因组学,metagenomics)是一种以环境样品中的微生物群体基因组为研究对象,以功能基因筛选和/或测序分析为研究手段,以微生物多样性、种群结构、进化关系、功能活性、相互协作关系及与环境之间的关系为研究目的的新的微生物研究方法。一般包括从环境样品中提取基因组DNA, 进行高通量测序分析,或克隆DNA到合适的载体,导入宿主菌体,筛选目的转化子等工作。
特定生物种基因组研究使人们的认识单元实现了从单一基因到基因集合的转变,宏基因组研究将使人们摆脱物种界限,揭示更高更复杂层次上的生命运动规律。在目前的基因结构功能认识和基因操作技术背景下,细菌宏基因组成为研究和开发的主要对象。细菌宏基因组、细菌人工染色体文库筛选和基因系统学分析使研究者能更有效地开发细菌基因资源,更深入地洞察细菌多样性。
宏基因组学在微生态学上的应用
Zhang 等构建红树林淤泥宏基因组文库, 通过PCR 扩增及变性梯度凝胶电泳(DGGE)对该区域固氮菌的多样性进行分析, 结果揭示红树林地区固氮菌的生物多样性特征, 其结果表明多数为变形菌, 也含少数的固氮菌属、除硫单胞菌属、德克斯氏菌属和根瘤菌等。
张薇等采用宏基因组技术对西北黄土高原柠条种植区土壤微生物多样性进行分析, 发现变形杆菌纲是根表土壤区系中的有优势微生物菌群(70.3%), 尤其存在大量能够诱导植物形成根瘤的根瘤菌和对植物有促生长作用的γ-Proteobacteria 类微生物, 说明了植物根系和土壤环境微生物菌群具有相互选择性。
Fierer 等通过构建牧场、沙漠、雨林土壤宏基因组文库对环境中细菌、古生菌、真菌及病毒多样性进行了研究, 并揭示了土壤环境中包含着大量不可培养的新病毒种类, 其基因型特征与常规培养获得的病毒具有很大的差异性。
Kim 等通过构建稻田土壤宏基因组文库, 利用多重置换扩增(MDA)技术对土壤样品中病毒基因多样性进行了研究, 结果表明扩增得到的病毒基因序列与目前报道的病毒序列具有很大的差异性, 进一步说明了土壤环境中包含着大量不可培养的病毒种类。
2003 年Breitbart 等首次通过构建宏基因组文库对人体排泄物中的未培养病毒多样性进行研究, 经过扩增及鸟枪测序鉴定,结果表明获得的病毒大约有1200 种基因型, 其基因序列与先前报道的病毒序列具有很大的差异性, 多为新的基因型病毒, 并揭示存在人体中的新病毒与人类疾病可能具有一定的相互性。
2005 年Cann 等通过构建宏基因组文库对马排泄物中的未培养病毒多样性进行研究, 经过测序鉴定, 获得233 种不同基因型的病毒, 其中52%为长尾噬菌体科, 26%为未分类的噬菌体, 17%为肌病毒科, 4%为短尾病毒科,2%为脊椎动物正痘病毒。
宏基因组学在海洋微生物资源开发上的应用
宏基因组工程与海洋生物学进行有机的结合,促使人类了解许多为培养海洋微生物的基因组序列及其功能产物,在海洋天然药物研究、海洋极端环境微生物研究、海洋微生物多样性探索中具有十分重要的应用前景。
Martín 等构建地中海深层水体微生物宏基因组文库, 通过序列分析和16S rRNA 系统发育比对,发现该水体的微生物种群与太平洋阿罗哈水域中层水体的微生物种群具有一定的相似性, 并提出在无光的条件下, 温度是影响微生物种群在水体中分布的主要因素。
2008 年肖凯等以宏基因组DNA 为模板, 采用不同的PCR 引物对温泉的高温水底沉积物微生物多样性进行分析, 发现了一株新的菌株JS-X2 与在美国黄石公园温泉发现的未培养细菌有95%的相似性, 并且与嗜热蓝细菌聚球藻有89%的相似性。
Sabet 等通过构建宏基因组文库对美国莫诺湖水体中噬菌体的多样性进行了研究, 研究发现不可培养的噬菌体才是该特殊生境中的优势群体, 揭示了海洋是一个巨大的未知RNA 病毒库。
Breitbart 等通过构建海水及海底沉积物宏基因组文库对该地区不可培养病毒的多样性进行分析, 结果发现扩增得到的病毒基因型中65%为新的基因型, 其中包含一类海藻病毒,多数病毒具有新的基因型, 与节肢动物和高等植物病毒存在很大的序列差异性。
宏基因组学在环境保护和污染修复上的应用
挖掘降解基因和功能菌株,进行生物修复
获取任何序列的基因或功能,由此合成新物质或发现新的生物物种。
发掘极端环境为生物的新物种,了解其耐受机制,帮助极端环境的污染修复。
从宏基因组中分离的重要基因元件组编成具有其他活性成分、或可降解污染物功能的基因簇,以替代原有不易降解化合物,或直接降解环境中石油烃、有害有害化合物、重金属。
可以有效地从环境中分离新的基因、化合物和生物催化剂;
所构建的工程菌可用于处理各种复杂污染物,是非常有前景的降解酶系基因筛选方法;
所获取的多样性信息可以在废水处理的各种反应器系统、污染物降解过程中微生物的作用和调控、营养物循环和富营养作用的微生物生态、微生物对环境和气候的监测等研究中发挥作用;
分析微生物种群多样性,检测评价环境健康。
宏基因组学在医学领域的应用
宏基因组技术的出现为新药物的探索和发现提供了可能的技术支持, 并扩大了微生物代谢产物及分子活性物质筛选平台。例如早在2000 年, Wang 等构建土壤宏基因组文库, 通过文库筛选获得TerragineA 及其相关成分, 目前已广泛应用于医学治疗领域, 证明了自然环境中的丰富微生物代谢产物可以通过宏基因组技术为人们所利用; 同年Brady 等从土壤宏基因组文库中筛选发现一种长链N-酰氨基酸抗生素物质; 并在2004 年构建凤梨科植物树茎流出液宏基因组文库, 筛选鉴定获得了抗菌物质PalmitoylPutrescine。2001 年Macneil等构建了土壤宏基因组BAC 文库, 通过序列分析筛选获得5 个能产生抗菌小分子物质靛玉红并对其相关成分进行研究; 2002 年Gillespie 等构建土壤宏基因组文库筛选获得两种抗菌物质Turbomycin A和B, 并且发现Turbomycin A 和B 对革兰氏阴性和革兰氏阳性菌具有广谱抗菌活性; 2003 年DiazTorres 等通过构建人唾液宏基因组文库, 筛选获得一种新的四环素抗性基因Tet, 该活性物质对四环素具有很好的抗性; 2008 年Mori 等通过活性污泥宏基因组文库筛选获得两种不同的博来霉素抗性基因, 经过比对发现于来源放射菌类基因差异较大, 可能为新的博来霉素抗性基因。
国内在利用宏基因组技术获取新型药物的研究较少, 尚处于萌芽阶段, 赵晶等从南极中山站排污口采集污泥,构建宏基因组文库, 并通过差异性DNA 修复实验(DDRT)筛选得到具有抗肿瘤效应的物质。同时利用宏基因组技术研究探讨人类肠道中不可培养微生物多样性也有了很大进展。如Kurokawa 等利用宏基因组技术对13 个处于不同年龄层的健康人体粪便微生物种群进行了研究, 结果分析表明未断奶婴儿的肠道微生物种群在系统发育和基因组成上具有较大个体差异性, 而在成人及已断奶儿童则呈现出高度的功能一致性, 并对成人及婴儿肠道内编码该生境微生物主要功能的基因家族的特性进行分析, 发现了一个新的人类肠道微生物基因家族和一个共扼转座子。2003 年Breitbart 等通过构建宏基因组文库对人体排放物中的未培养病毒多样性进行研究,经过鸟枪测序法鉴定, 获得的病毒大约有1200 种基因型, 结果比对表明其基因序列与先前报道的病毒具有很大的差异性, 大多数为新病毒, 并证明这些病毒极有可能与人类的疾病有着密切的关系。2008 年Finkbeiner 等通过构建12 个腹泻小孩肠道内容物宏基因组文库, 来观察病人肠道中病毒的生物多样性, 发现扩增得到的病毒序列与GenBank 病毒库中的已知序列同源性很低, 并推断这些病毒极有可能与人类的腹泻疾病有着密切的关系。随着宏基因组技术的成熟, 必将加快宏基因组技术在医学中的应用。在人体微生物抗药性的研究, 人体与不可培养病原菌的相互关系的探索等方面将做出重大贡献。
宏基因组学在生物酶制剂开发中的应用
宏基因组学技术最引人注目的贡献主要集中在新型生物酶制剂的探索和开发领域。传统的新型酶的筛选方法大大限制了筛选的广泛性和有效性。宏基因组学通过直接从环境中提取DNA 样品,尽可能为后面的筛选提供更加全面和多样的基因资源,从而有效地提高了新酶的筛选效率。
近年来研究者们已成功构建了土壤、海底淤泥、温泉淤泥、油厂污泥、动物瘤胃内容物、动物粪便等宏基因组文库,并筛选到脂肪酶、蛋白酶、淀粉酶、乙醇氧化酶、木聚糖酶、纤维素酶及脱羧酶等酶制剂, 并且在此基础上获得新酶的许多特征信息。所采用的载体种类十分广泛, 包括Fosmid、Cosmid、BAC、λ噬菌体以及各种穿梭载体, 所采用的宿主系统为常用的大肠杆菌、链霉菌和假单胞菌等。通过宏基因文库筛选得到的生物分子大多数与已知的基因产物相似性差或者完全是新的分子, 这些新的生物分子主要来源于环境未培养微生物的基因和其多样的代谢物。
环境样品DNA 的克隆和筛选只是所有环境遗传信息多样性的一小部分, 环境微生物和宏基因组的多样性仍旧是发现新的天然活性产物的丰富广阔资源, 为研究者们探索开发新的生物催化剂提供了巨大的资源空间。