
技术专题
基因组学最新研究进展
1. Nature:揭示出人基因组的古老弱点
doi:10.1038/nature24018

在一项新的研究中,来自冰岛基因解码公司(deCODE genetics)、冰岛大学和雷克雅未克大学的研究人员利用来自冰岛人口中的1.4万人(包括1500个三人组,每个三人组为父母及其孩子)的全基因组数据,针对人类中的序列多样性如何是性别、年龄和在基因组中的突变类型和位置之间在进化过程中相互作用的结果提供了迄今为止最详细的描述。相关研究结果于2017年9月20日在线发表在Nature期刊上,论文标题为“Parental influence on human germline de novo mutations in 1,548 trios from Iceland”。
论文通信作者、基因解码公司首席执行官Kari Stefansson说,“这是我们正在开展的研究的一个重要的新篇章,有助理解促进基因组多样性产生和在此过程中促进我们的物种进化的机制。新的突变为进化的环境提供了一种重要的部分,从而使得人类基因组的新版本不断流入到环境中。然而,它们也被认为是导致大多数罕见的儿童疾病病例的罪魁祸首。因此,为整个人群提供这些突变的完整目录不仅在科学上是激动人心的,而且也为改善罕见疾病诊断作出重要的贡献。”
这项新的研究表明在大约10%的基因组区域中,来自母亲的新突变与来自父亲的新突变在数量上是相同的。在这些区域中,大多数源自母亲的新突变是C>G突变,而且是起源自DNA双链断裂修复存在的缺陷。在这种10%的基因组区域中,C>G SNP(单核苷酸多态性)的密度也是非常高的,不过在不太常见的等位基因中出现的次数存在着显著的变化。这表明这种基因组区域在非常长的时间里容易发生DNA双链断裂。这种相同的基因组区域也在黑猩猩(chimpanzees)中表现出较高的C>G SNP密度,在大猩猩(gorillas)中具有较低的密度,而在猩猩(orangutans)中则一点儿都不高。
Stefansson补充道,“在这种10%的基因组区域中,新突变出现的频率几乎是剩余的基因组中的两倍。显然地,这种区域中的基因赋予我们的属性必须比其他的属性进化得更快。”
这项研究使用了基因解码公司在冰岛的独特遗传资源,并且是建立在将父母的年龄和性别与重组变异性和多种突变类型之间相关联在一起的早期结果的基础之上的。之所以被称作新突变是因为它们是在卵子和精子形成期间产生的。这些研究人员能够为这些将来自上千人和家庭几代人的全基因组序列数据与他们的孩子出生时的父母年龄信息关联在一起的新突变开发出如此庞大的数据集和目录。
2. Nature:我国科学家成功对深圳拟兰基因组进行测序,揭示其进化之谜
doi:10.1038/nature23897

在一项新的研究中,来自中国深圳市兰科植物保护研究中心、中科院植物研究所、华南农业大学、武汉大众源生科技服务有限公司、福建农林大学、清华大学深圳研究生院、中国台湾国立成功大学;比利时根特大学、VIB植物系统生物学中心;日本国家农业与食品研究组织花卉园艺学研究所、国家先进工业科学技术研究所、大成建设株式会社、埼玉大学;南非基因组学研究院的研究人员对兰科植物深圳拟兰(Apostasia shenzhenica)的基因组进行了测序。他们描述了他们采用的测序方法以及在这个过程中他们学到了什么。相关研究结果于2017年9月13日在线发表在Nature期刊上,论文标题为“The Apostasia genome and the evolution of orchids”。
说兰科植物在当今世界流行是一种轻描淡写的说法,正如这些研究人员注意到的那样,它们当前大约占所有开花植物的10%,而且在地球上除大多数极端的栖息地之外的地方生长。鉴于它具有较强的迁移和适应不同条件的能力,多年来它就已激起了科学家们的兴趣。在这项新的研究中,这些研究人员着重关注深圳拟兰,即一种在中国南方发现的兰科植物。它大部分是绿色的,开出黄色的花。了解深圳拟兰花朵的基因组组成揭示出它发生进化从而非常好地适应新条件的方式。
这些研究人员报道他们利用10X基因组学平台(genomics scaffolding)开展短读取测序和长读取测序,从而获得深圳拟兰的基因组序列。他们也报道他们利用他们的研究结果对这种植物与其他的兰科植物进行比较,并且基于转录组数据找出相同之处和不同之处。
这些研究人员为深圳拟兰属于兰科植物提供强大的证据:它的大部分基因组几乎是其他的兰科植物基因组的虚拟拷贝。他们也发现,正如之前猜测的那样,它在几百万年前与其他的兰科植物区分开来。他们还发现在兰科植物出现差异和随后进化出5种亚科之后,它们经历一段重大的灭绝时期。他们指出正是在这个时期,诸如知名的“唇状物(lip)”之类的特征产生了。
这些研究人员的发现提示着深圳拟兰是进一步开展研究的一种较好的候选对象,这是因为它可能提供进化线索,比如一些导致兰科植物产生大量花粉(pollonia)和比其他的植物更轻的种子的因素。它也可能解释了另一种知名的特征:利用其他的植物提供支持的能力。(生物谷 Bioon.com)
3. Cell Systems:大型基因组数据库分析或能帮助寻找和未来疾病发病风险相关的遗传突变
DOI: 10.1016/j.cels.2017.06.016

如今,临床医生们会越来越多地使用遗传测试技术来寻找罕见疾病患者的发病原因,但寻找到病因仅仅是我们对抗疾病战斗的一部分,基因组分析的下一个前沿领域就是预测遗传突变是否是引发疾病的先兆。近日,一项刊登在国际杂志Cell Systems上的研究报告中,来自哈佛医学院的研究人员通过研究开发出了一种新方法,这种方法能够增加研究人员预测遗传突变作为疾病病因的准确率;进行大规模的基因组分析后,研究人员或能有效预测并不是很清楚的致病突变到底是如何引发机体疾病症状的,同时还能够帮助评估患者在后期出现临床疾病的可能性。
这项研究中,研究人员重点对罕见的遗传性疾病进行了研究,因为这些疾病很容易同根源性致病突变相匹配,为了阐明多个患者机体中出现的突变,并且将突变同特殊疾病相联系起来,研究人员必须对相同疾病患者的大量基因组测序数据进行观察分析,随后他们将患者的基因组数据同未患病个体机体的基因组数据对比。
文章中,研究人员检测了一种假设,即影响器官功能的遗传性疾病很有可能源于组成器官的组织中大量表达的基因发生突变所致;比如,如果一个特定的基因能够产生一种在心脏中普遍存在的蛋白质,那么该基因的突变很有可能会引发患者出现遗传性的心脏疾病,当然这一概念本身并不新鲜,但此前研究中研究人员或许得出了并不一致的结论。
研究者对不同组织中相同基因的表达情况进行了对比,同时还研究了相同组织中不同基因的表达情况;当然,这种假设在某些组织中存在但在其它组织中似乎并不存在;研究者发现,在心脏、大脑、皮肤和肌肉中高度表达的基因发生突变或许会经常引发这些器官患病,然而,在乳腺、甲状腺和胃部组织中,基因的表达水平和疾病发生风险并无关联,相关研究结果表明,在某些组织中或许存在一种较为复杂的互作关系。
研究者Isaac Kohane说道,本文研究阐明了利用大数据来鉴别基因表达模式的重要性,这或许能帮助我们理解遗传突变发生的机制,同时相关研究结果还能够给研究人员提供线索,帮助他们监测一个器官出现症状但却含有和多个器官有关的突变的患者,比如,遗传性肌肉疾病背后的突变或许被发现会引发一小部分患者出现神经发育性的障碍。最后研究者Feiglin表示,这些巨大数据库的美妙之处在于我们可以利用现有的数据做出更多新的发现。
4. PNAS:重磅!科学家成功利用全基因组测序数据对人群进行鉴别
doi: 10.1073/pnas.1711125114

近日,一项刊登在国际杂志Proceedings of the National Academy of Sciences上的研究报告中,来自国外的研究人员通过研究,利用全基因组测序和机器学习等技术成功实现了对每个人面部及其它机体特征进行鉴别。
研究人员认为,这项研究或为法医提供了新的方法来识别凶手,同时本文研究对于数据保密、识别以及个体的充分知情同意也具有重要的意义。随着越来越多基因组数据的产生以及公共数据库的扩大,我们往往需要进行更多的公众审议。
这项研究中,研究人员对1061名年龄在18-82岁不同种族的人群进行研究,对参与者进行了至少深入为30X的基因组测序,同时研究人员还收集了参与者的一些表型数据,包括3D面部图像、声音样本、眼睛和肤色、年龄、身高及体重等信息。研究人员能够准确预测参与者的眼睛颜色、肤色及型别,但预测其它更复杂的遗传特点似乎比较困难,但他们认为他们所开发的预测模式是合理的,但后期还需要大量的研究对象才能够进行更为精准的预测。
此外,研究人员还开发了一种名为最大熵算法的机器学习算法,这种算法能够在所有的预测模型中帮助寻找最佳的组合来将全基因组测序数据同表型和人口数据进行匹配,来进行更为准确的人群识别,平均而言在不同种族中能达到80%的识别率,在非洲裔美国人和欧洲参与者中能够达到50%的识别率。
研究者Venter表示,我们进行这项研究是为了证明我们机体的基因组能够编码任何制造机体的元件,尽管这是对一个有限的群体进行的概念验证研究,但我们认为随着这项研究以及我们所开发的数据库中参与者数量的增加,我们就能够更加准确地通过个体的基因组及其它信息进行人群的准确识别。
Venter补充道,此外我们都非常关心,目前公众和很多研究团体对于个体在基因组方面的隐私和安全并没有过多的关注,因此这就要求我们进行我们后期应该进行更多深入的分析,并且开发出更多新型的技术解决方案。本文研究阐明了新型成像技术对大规模人群进行特征筛选的潜力,机器学习技术能够进行完全自动化的数据解释,同时在科学研究中也扮演着关键的角色。
5. PLoS ONE:新技术或能实现在人类“垃圾基因组”中搜寻引发疾病的关键突变
doi: 10.1371/journal.pone.0181604

当医生无法诊断患者的病情时,他们往往会寻找“遗传侦探”,如今基因组测序技术使用还没有10年时间,但其常常能够通过对患者机体的DNA进行搜索来寻找引发多种神秘疾病的突变。尽管取得了一系列成功,但基因组测序技术经常会得到空白结果,实际上诱发疾病的突变仅能够在三分之一至四分之一且机体存在强烈遗传状况的患者中存在。
为什么很多研究人员会得到空白结果呢?原因就在于“黑暗基因组”,目前科学家们仅对人类2%的基因组进行了解析,这2%的基因组中包含有能够编码细胞所有蛋白的20000个基因,剩下98%的黑暗基因组对于科学家们而言仍然是个谜题,尽管有研究认为这些非编码的基因组能够调节基因的开启和关闭,但具体细节研究者却不得而知。
近日,一项刊登在国际杂志PLoS ONE上的研究报告中,来自哥伦比亚大学医学中心的研究人员通过研究试图在巨大的非编码基因组中寻找诱发疾病的致病性突变,文章中,研究人员开发了一种名为Orion的新技术,该技术能够通过在人类基因组中进行搜寻来帮助标记可能含有促疾病发生的遗传改变的非编码基因组区域。
研究者Goldstein说道,我们能够利用Orion技术来帮助在患者机体中寻找测序技术无法找到的致病突变,Orion技术能够对1662名个体机体中的整个基因组进行对比分析,鉴别出人群之间发生微小改变的DNA片段,由于这些区域无法忍受改变,因此这对于研究人员后期研究至关重要;也就是说,相比耐受区域而言,不耐受区域所发生的突变很有可能会引发疾病,随后当研究人员对所鉴别出的非编码突变的位点进行图谱绘制时他们证实了该预测,在Orion所鉴别的不耐受区域中往往会存在更多突变。
此前深入探究非编码基因组的研究方法重点对多个物种机体中固定不变的非编码基因组进行分析,然而本文研究中研究人员所开发的Orion技术则能够鉴别出人类基因组中发挥重要新功能的特殊区域。Goldstein表示,Orion技术目前的开发并未彻底完成,随着更多基因组区域被测序,Orion搜索区域的分辨率也会明显改善。
最后研究者表示,目前我们对于Orion技术表示非常乐观,其将会作为一种有用的工具来帮助鉴别基因组中会影响罕见和常见疾病发生风险的基因突变。
6. Science:基因组测序表明玉米在几千年前适应北美高原地区
doi:10.1126/science.aam9425

在一项新的研究中,来自美国、德国和墨西哥的研究人员发现证据表明玉米在几千年前通过进化适应了美国西南高地(或者说高原地区)。他们概述了他们的基因组研究,揭示出允许这种植物在更加恶劣的环境中存活的遗传变化。相关研究结果发表在2017年8月4日的Science期刊上,论文标题为“Genomic estimation of complex traits reveals ancient maize adaptation to temperate North America”。
玉米起源自墨西哥,大约在4000年前到达美国西南低地。通过这样做,它快速地成为北美洲的最为重要的农作物之一。但是,正如这些研究人员注意到的那样,它在此后的2000年的时间内并没有到达美国西南高地。这让考古学家感到困惑。为了更好地理解这种延迟为何发生,这些研究人员研究了1970年代在犹他州高地的一个洞穴中发现的2000前的玉米棒(maize cob)样品。
为了更多地了解物理特征,这些研究人员对15个玉米棒的基因组进行测序,并且将获得的结果与其他的玉米品系进行比较。他们报道在这个洞穴周围的玉米植物并不如在较低海拔的地方生长的其他玉米植物那么高,而且具有更多的分枝,换言之,他们描述这些玉米植物要比其他的玉米植物更加茂密,这一特征允许它们在更加寒冷的地方茁壮生长。他们也发现证据表明这些玉米植物要比大多数其他的玉米植物更早地开花,这一特征有助它们在较早的霜冻在更高海拔的地方出现之前产生种子。
当这些玉米植物通过进化适应这种更加恶劣的环境时,生活在那里的早期人类已开始将玉米作为他们的食物,正如美国西南低地的早期人类早在几千年前所做的那样。
这些研究人员指出更好地了解玉米如何经过进化适应更加寒冷的高地会提供重要的见解,这是因为气候变化迫使很多农作物为了适应新的环境条件而做同样的事情。他们也注意到一种富含类胡萝卜素的玉米在美国西南地区而不在墨西哥进化出来。这种富含类胡萝卜素的玉米如今被用来做爆米花。
7. Nat Methods:新型基因组工具CITE-seq或能实现单细胞大规模多维度分析
doi:10.1038/nmeth.4380

近日,一项刊登在国际杂志Nature Methods上的研究报告中,来自纽约基因组研究中心(NYGC)的研究人员通过研究开发了一种新技术,或能帮助推动单细胞RNA测序的进程,单细胞RNA测序是基因组研究的重要领域,其能够帮助研究人员深入解读单细胞的特性,同时还能够帮助有效区分不同的细胞类型,以及在单细胞水平下研究多种人类疾病的发病机制。
这种名为CITE-seq(Cellular Indexing of Transcriptomes and Epitopes by sequencing,通过测序来进行转录组和表位的细胞索引技术)的新型测序技术能够对数千个单细胞的表面蛋白标记物进行测定,同时还能够对相同单细胞中的信使RNA进行测序。如今研究人员正在对该技术进行概念验证研究,他们结合转录组学技术,对8000个单细胞表面的10种表面蛋白进行了监测,截至目前为止,这项研究是对单细胞进行的最大规模且多维度的分析。
研究者Marlon Stoeckius博士表示,并没有其它方法能够让我们在同一尺度下同时对细胞的蛋白质及转录组学特性进行测定;而CITE-seq已经能够为转录组学分析建立新的方法,同时并不会对所产生的数据的质量产生不利的影响。此前的研究方法主要依赖于在进行单细胞RNA测序之前利用细胞计数法来捕捉单细胞的蛋白质信息,而当前的方法吞吐量较低,而且受限于相对较少的蛋白质标记物。
CITE-seq技术的蛋白质检测组分基于DNA条形码抗体,其能够产生排序的读数,而且还能够同时将细胞的转录组学信息捕获,该技术所产生的蛋白质和RNA数据的整合通常需要进行定制化的数据分析。研究者表示,以CITE-seq技术为例进行研究,我们就能够利用多通道数据来鉴别出自然杀伤细胞(NK细胞)的亚群,而通常仅利用转录组学研究很难对自然杀伤细胞的亚群进行区分。
CITE-seq技术能够精细化分析细胞群体的能力将会使其在研究领域具有更多的应用价值;最后研究者Stoeckius说道,未来一种可能性的研究方向就是在肿瘤样本中利用CITE-seq技术来检测单一的肿瘤细胞以及能够浸润肿瘤组织的多种不同的免疫细胞池;这项新技术或能用于对肿瘤异质性的深度特性分析,当然其或许还能够推动免疫治疗领域的进展,比如新型肿瘤免疫疗法的开发等。
8. Nature:首次对成神经管细胞瘤进行全基因组分析
doi:10.1038/nature22973
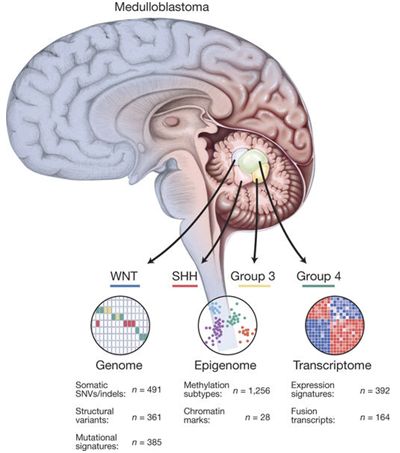
在一项新的划时代研究中,来自德国、加拿大、美国、荷兰、丹麦、俄罗斯、西班牙和日本的研究人员对成神经管细胞瘤(medulloblastoma)开展迄今为止最为全面的分析,鉴定出导致75%以上的成神经管细胞瘤的基因组变化,包括仅在这个最少了解的脑瘤亚群中发现的两个新的潜在癌基因。相关研究结果于2017年7月19日在线发表在Nature期刊上,论文标题为“The whole-genome landscape of medulloblastoma subtypes”。
对来自将近500名患者的肿瘤和正常组织开展的综合基因组分析结果揭示出新的突变和遗传错误。这些发现将有助开发迫切需要的精准疗法。这些靶向疗法旨在增加存活率,同时降低治疗相关的副作用。
论文共同第一作者、美国圣犹大儿童研究医院发育神经生物学科研究员Paul Northcott博士说,“我们的目标是在分子水平上理解每名患者的肿瘤以便更好地开发出个人化疗法。这项研究为全世界的神经肿瘤学研究人员和医生提供一种丰富的资源进行挖掘以便更好地理解和靶向这些肿瘤。”
成神经管细胞瘤是最为常见的恶性儿童脑瘤。在美国,每年有250~500人被诊断患上这种脑瘤;大多数患者小于16岁。成神经管细胞瘤存在4种主要的亚型:WNT、SHH(Sonic Hedgehog, 音猬因子)、III型(Group 3)和IV型(Group 4)。这些亚型是由不同的遗传变化导致的,起源于不同的细胞,而且经常表现出不同的临床结果。大约95%的WNT亚型肿瘤患者成为长期的存活者,而对于III型肿瘤患者而言,这一数字大约是50%。当前还没有针对III型肿瘤和IV型肿瘤的靶向疗法,这两种肿瘤亚型占成神经管细胞瘤病例的65%~70%。
这项研究是针对任何一种肿瘤开展的最大全基因组测序研究之一。全基因组测序或全外显子测序是在从491名确诊的成神经管细胞瘤患者体内收集的肿瘤和正常组织中开展的。
这些研究人员也分析了来自1256名成神经管细胞瘤患者的DNA甲基化数据和补充性的表观遗传数据,和来自392种肿瘤样品的基因表达数据。
Northcott说,“结合更好的分子工具对基因数据、表观遗传数据和转录数据进行分类,这会导致在这种高度复杂和多变的癌症中之前被遗漏掉的发现。”论文共同作者、圣犹大儿童研究医院儿科医学主任Amar Gajjar博士补充道,“利用先进的基因组技术对导致成神经管细胞瘤的遗传异常获得新的认识对开发相关的模型来测试靶向疗法是至关重要的。”
这些发现包括鉴定出两个潜在新的癌基因:KBTBD4和PRDM6。这两个基因仅在III型和IV型成神经管细胞瘤中发现到,而且之前并未发现与任何一种癌症相关联。
KBTBD4是在这两种成神经管细胞瘤亚型中最常见的发生突变的基因。这种突变涉及将一到两个氨基酸插入到这个基因的“热点(hot spot)”区域中。据预测,KBTBD4蛋白与标记其他蛋白以便细胞循环利用的泛素连接酶一起发挥功能,但是Northcott说,更好地理解这种正常的蛋白和发生突变的蛋白如何发挥功能的研究正在开展中。
PRDM6是一种对基因活性进行表观遗传调节的基因,它的表达在IV型成神经管细胞瘤中显著增加。有证据提示着这种增加的活性是由于DNA重排导致的“增强子劫持(enhancer hijacking)”,即重新配置高度有活性的增强子来激活PRDM6表达。
PRDM6激活导致17%的IV型成神经管细胞瘤。如果PRDM6经证实是一种癌基因的话,那么研究人员希望这将会导致首个IV型成神经管细胞瘤实验室模型。这样的模型在临床进展中发挥着一种至关重要的作用。IV型成神经管细胞瘤大约占成神经管细胞瘤的40%。
总而言之,这项综合分析鉴定出导致75%以上的成神经管细胞瘤的基因变化和细胞通路。在这项研究之前,科学家们仅鉴定出III型和IV型成神经管细胞瘤驱动突变的三分之一不到。这些数据也揭示出不同成神经管细胞瘤亚型(特别是III型和IV型)的额外复杂性。Gajjar说,“这项研究提供的更加精致的成神经管细胞瘤分类为对改进的患者风险分层和开发靶向导致他们的疾病产生的基因组变化的个人化疗法开辟新的途径。
9. Science:首次对野生二粒小麦进行基因组测序,有助改进未来的小麦产量和安全性
doi:10.1126/science.aan0032

在一项新的研究中,一个国际团队有史以来首次发布野生二粒小麦(Wild Emmer wheat)的基因组序列。相关研究结果发表在2017年7月7日的Science期刊上,论文标题为“Wild emmer genome architecture and diversity elucidate wheat evolution and domestication”。论文通信作者为以色列特拉维夫大学植物科学与食品安全学院研究员Assaf Distelfeld博士。
野生二粒小麦是世界上几乎所有驯化小麦(包括硬质小麦和面包小麦)的原始形态。它具有太低的产量而不被农民们种植,但是它含有很多吸引人的特征。这些特征正在被植物育种者用来改进小麦品种。
论文共同作者、加拿大萨斯喀彻温大学研究员Curtis Pozniak博士说,“我们如此快地获得野生二粒小麦基因组序列的能力是基因组研究向前迈出的一大步。小麦提供全世界人们消化的卡路里的将近20%,因此强烈地关注对小麦的产量和品质的关注对我们未来的食物供应是至关重要的。”
Distelfeld博士说,“从生物学和历史的角度来看,我们为我们能够用来研究农业出现之前的小麦创建一种‘时光隧道(time tunnel)’。通过与现代小麦相比,我们能够鉴定出参与驯化的基因。驯化指的是将在野外生长的小麦转化为现代的小麦品种。尽管野生小麦的种子容易掉落和扩散,但是在驯化小麦中,两个基因发生变化,从而使得种子仍然附着到小麦茎秆上;正是这种特征使得人类能够收割小麦。”
论文共同作者、美国堪萨斯州立大学研究员Eduard Akhunov博士说,“这种新的资源允许我们鉴定出许多控制着早期人类在小麦驯化期间选择的主要性状的其他基因,它们可作为开发现代小麦品种的基础。这些基因为能够在未来开展育种工作提供一种宝贵的资源。野生二粒小麦能够有助改进小麦籽粒的营养品质,以及对疾病和水受限的条件的耐受性。”
论文共同作者、以色列耶路撒冷希伯来大学研究员Zvi Peleg博士解释道,“新的基因组工具已正在被用来鉴定在变化的环境下改善小麦产量的新基因。尽管很多现代小麦品种容易遭受水胁迫,但是野生二粒小麦已在容易发生干旱的地中海气候下经历着比较长的进化历史。因此,在小麦培育程序中使用野生小麦的基因会提高在缺水条件下的产量。”
论文共同作者、NRGene公司首席执行官Gil Ronen博士说,“小麦基因组比大多数其他的作物更加复杂,而且它的基因组是人类的基因组大小的4倍。不过,我们开发的计算技术允许我们快读地组装在野生二粒小麦的14条染色体中发现的非常大的和复杂的基因组。这在之前的基因组研究中是无法实现的。”
这些研究人员首次让野生二粒小麦的14条染色体的基因组序列以一种优化的次序进行组装。德国莱布尼茨植物遗传学与农作物研究所遗传资源基因组学主任Nils Stein博士说,“这一点最初在人类中测试过,最近在大麦中证实过。人类和大麦的基因组都比野生二粒小麦要小。这些创新性的技术引发大型谷类植物基因组组装变革。”
论文共同作者、意大利农业研究与经济委员会(Council of Agricultural Research and Economics, CREA)基因组学与生物信息学研究中心主任Luigi Cattivelli博士说,“用于野生二粒小麦的这种测序方法是史无前例的,为对硬质小麦(野生二粒小麦的一种驯化形式)进行基因组测序铺平道路。如今,我们能够更好地理解人类如何将这种野生的植物转化为一种现代的高产量的高质量的作物。这种野生二粒小麦测序方法是对整个小麦研究界作出的一种宝贵的贡献,有助改进和更好地理解小麦营养机理。”
Distelfeld博士作出结论,“我们如今有这些工具来直接地研究作物和比之前更加高效地利用我们的发现。”